Loading required package: carData
Attaching package: 'car'
The following object is masked from 'package:dplyr':
recode
The following object is masked from 'package:purrr':
some
Data below was collected from a pilot study conducted in Autumn 2023 where field-collected individual Botryllus schlosseri were exposed to a low and high treatment of nickel chloride and then were dissected for primary buds. The objective was to evaluate if a pre-nickel exposure in Botryllus improved primary epithelial cell monolayer proliferation. Refer to Pilot 1.1 Nickel Exposure notebook post for experimental design details.
Retrieving Data from Google Sheets
Note, if you do this, make sure your spreadsheet is publicly available to anyone with a link.
During the study, we documented blastogenic stage with the 7 level blastogenic stage scale For simplicity in our graphs and our interpretation, we will be being the stages into a 4 level blastogenic stage scale
# Create a new column 'simple_stage' based on conditionscell <- cell %>%mutate(simple_stage =case_when( stage_at_dissection %in%c("A1", "A2") ~"A", stage_at_dissection %in%c("B1", "B2") ~"B", stage_at_dissection %in%c("C1", "C2") ~"C", stage_at_dissection =="TO"~"TO",TRUE~NA_character_# This will handle any other cases or return NA if none match ))
Cleaning up and subsetting data.
cell$date <-mdy(cell$date) #convert the date column from characters to true datecell_pilot <- cell[cell$experiment ==0, ] # only want the pilot experiment, experiment 1 failed in poor dissecting technique# Subset the 'cell_pilot' data frame to include rows with hours post seeding (hps) values of 0, 18, 38, and 70 hps.cell_pilot2 <- cell_pilot[cell_pilot$hps %in%c(0, 18, 38, 70), ]# Set the treatment, animal ID, and date as factors.cell_fact <- cell_pilot2 %>%mutate(treatment_mg_per_L =as.factor(treatment_mg_per_L)) %>%mutate(animal_ID =as.factor(animal_ID)) %>%mutate(date =as.factor(date))summary(cell_fact)
date experiment plate_Number well_Placements
2023-09-30:12 Min. :0 Min. :1 Length:48
2023-10-01:12 1st Qu.:0 1st Qu.:1 Class :character
2023-10-02:12 Median :0 Median :1 Mode :character
2023-10-03:12 Mean :0 Mean :1
3rd Qu.:0 3rd Qu.:1
Max. :0 Max. :1
treatment_mg_per_L replicate animal_ID stage_2
0 :16 Length:48 S147C001: 4 Length:48
5 :16 Class :character S148C001: 4 Class :character
45:16 Mode :character S149C001: 4 Mode :character
S151C001: 4
S154C001: 4
S158C001: 4
(Other) :24
stage_at_dissection initials hps no_attached_tissue
Length:48 Length:48 Min. : 0.0 Min. : 1.000
Class :character Class :character 1st Qu.:13.5 1st Qu.: 4.750
Mode :character Mode :character Median :28.0 Median : 5.000
Mean :31.5 Mean : 5.646
3rd Qu.:46.0 3rd Qu.: 7.000
Max. :70.0 Max. :10.000
no_mono_out image percent_media_replaced tissue_total
Min. :0.000 Length:48 Min. : 0 Min. : 2.000
1st Qu.:0.000 Class :character 1st Qu.: 0 1st Qu.: 5.000
Median :0.000 Mode :character Median : 0 Median : 7.000
Mean :0.375 Mean : 25 Mean : 6.583
3rd Qu.:1.000 3rd Qu.: 25 3rd Qu.: 8.000
Max. :1.000 Max. :100 Max. :10.000
prim_bud_no zooid_no float_prim float_zooid
Min. :0.000 Min. : 0.000 Min. :0.0000 Min. :0.0000
1st Qu.:1.500 1st Qu.: 0.000 1st Qu.:0.0000 1st Qu.:0.0000
Median :2.000 Median : 0.000 Median :1.0000 Median :0.0000
Mean :3.745 Mean : 2.574 Mean :0.7234 Mean :0.2766
3rd Qu.:6.000 3rd Qu.: 6.000 3rd Qu.:1.0000 3rd Qu.:1.0000
Max. :8.000 Max. :10.000 Max. :3.0000 Max. :1.0000
NA's :1 NA's :1 NA's :1 NA's :1
comments simple_stage
Length:48 Length:48
Class :character Class :character
Mode :character Mode :character
Calculate the percent of monolayer output per seeded tissue piece based on the first 12 rows of ‘attached_tissue’. Here we are calculating the amount of monolayers that occurred relative to the amount of originally seeded tissue. Overtime, seeded tissue detaches and is removed from the plate so if you were to calculate this ratio per day it would be off. If using this, make sure the data frame is in order from A1-C4 wells otherwise it’s miscalculated.
percent_output <- ((cell_fact$no_mono_out) /head(cell_fact$no_attached_tissue, 12)) *100# Add the newly calculated percent output values to the old data framecell_fact <-cbind(cell_fact,percent_output)# Create a new data frame 'cell_fact_filtered' excluding rows with 'C2' and 'C4' in 'stage_at_dissection'cell_fact_filtered <- cell_fact[!(cell_fact$well_Placements %in%c("C2")), ]
Summary Statistics
Number of tissue pieces attached:
# Calculate the mean attached tissuemean_tissue_attached <- cell_fact_filtered %>%group_by(hps, treatment_mg_per_L, simple_stage) %>%summarise(mean_attached =round(mean(no_attached_tissue, na.rm =TRUE)),num_points =n())
`summarise()` has grouped output by 'hps', 'treatment_mg_per_L'. You can
override using the `.groups` argument.
`summarise()` has grouped output by 'hps', 'treatment_mg_per_L'. You can
override using the `.groups` argument.
n <-4SE_tissue_attached <- SD_tissue_attached$SD_attached/sqrt(n)# Print the calculated mean health scoresprint(mean_tissue_attached)
# A tibble: 36 × 5
# Groups: hps, treatment_mg_per_L [12]
hps treatment_mg_per_L simple_stage mean_attached num_points
<int> <fct> <chr> <dbl> <int>
1 0 0 C 4 2
2 0 0 TO 7 1
3 0 5 B 5 1
4 0 5 C 8 2
5 0 5 TO 6 1
6 0 45 A 5 1
7 0 45 B 10 1
8 0 45 C 7 1
9 0 45 TO 5 1
10 18 0 C 4 2
# ℹ 26 more rows
print(SE_tissue_attached)
[1] 0.5 NA NA 0.5 NA NA NA NA NA 0.5 NA NA 0.5 NA NA NA NA NA 0.5
[20] NA NA 0.5 NA NA NA NA NA 0.5 NA NA 0.5 NA NA NA NA NA
Number of monolayer output:
# Calculate the mean mean_mono_out <- cell_fact_filtered %>%group_by(hps, treatment_mg_per_L, simple_stage) %>%summarise(no_mono =mean(no_mono_out, na.rm =TRUE),num_points =n())
`summarise()` has grouped output by 'hps', 'treatment_mg_per_L'. You can
override using the `.groups` argument.
# Calculate SD and filter out groups with fewer than 2 observationsSD_mono_out <- cell_fact_filtered %>%group_by(hps, treatment_mg_per_L, simple_stage) %>%summarise(SD_mono =sd(no_mono_out, na.rm =TRUE),num_points =n())
`summarise()` has grouped output by 'hps', 'treatment_mg_per_L'. You can
override using the `.groups` argument.
n <-4SE_mono <- SD_mono_out$SD_mono/sqrt(n)# Print the calculated mean health scoresprint(mean_mono_out)
# A tibble: 36 × 5
# Groups: hps, treatment_mg_per_L [12]
hps treatment_mg_per_L simple_stage no_mono num_points
<int> <fct> <chr> <dbl> <int>
1 0 0 C 0 2
2 0 0 TO 0 1
3 0 5 B 0 1
4 0 5 C 0 2
5 0 5 TO 0 1
6 0 45 A 0 1
7 0 45 B 0 1
8 0 45 C 0 1
9 0 45 TO 0 1
10 18 0 C 0 2
# ℹ 26 more rows
print(SE_mono)
[1] 0.0000000 NA NA 0.0000000 NA NA NA
[8] NA NA 0.0000000 NA NA 0.3535534 NA
[15] NA NA NA NA 0.0000000 NA NA
[22] 0.3535534 NA NA NA NA NA 0.0000000
[29] NA NA 0.3535534 NA NA NA NA
[36] NA
Monolayer output by relative tissue seeded (percent):
# Calculate the mean mean_ratio_mono_out <- cell_fact_filtered %>%group_by(hps, treatment_mg_per_L, simple_stage) %>%summarise(percent_mean_mono =mean(percent_output, na.rm =TRUE),num_points =n())
`summarise()` has grouped output by 'hps', 'treatment_mg_per_L'. You can
override using the `.groups` argument.
# Calculate SD and filter out groups with fewer than 2 observationsSD_ratio_mono_out <- cell_fact_filtered %>%group_by(hps, treatment_mg_per_L, simple_stage) %>%summarise(SD_ratio_mono =sd(percent_output, na.rm =TRUE),num_points =n())
`summarise()` has grouped output by 'hps', 'treatment_mg_per_L'. You can
override using the `.groups` argument.
n <-4SE_mono_ratio <- SD_ratio_mono_out$SD_ratio_mono/sqrt(n)# Print the calculated mean health scoresprint(mean_ratio_mono_out)
# A tibble: 36 × 5
# Groups: hps, treatment_mg_per_L [12]
hps treatment_mg_per_L simple_stage percent_mean_mono num_points
<int> <fct> <chr> <dbl> <int>
1 0 0 C 0 2
2 0 0 TO 0 1
3 0 5 B 0 1
4 0 5 C 0 2
5 0 5 TO 0 1
6 0 45 A 0 1
7 0 45 B 0 1
8 0 45 C 0 1
9 0 45 TO 0 1
10 18 0 C 0 2
# ℹ 26 more rows
print(SE_mono_ratio)
[1] 0.000000 NA NA 0.000000 NA NA NA NA
[9] NA 0.000000 NA NA 4.419417 NA NA NA
[17] NA NA 0.000000 NA NA 4.419417 NA NA
[25] NA NA NA 0.000000 NA NA 4.419417 NA
[33] NA NA NA NA
Graphs
Below are graphs used in the SICB poster generated. Graph settings are set to be really large for printing and don’t render well in this format so instead are figures with attached images of the produced graphs.
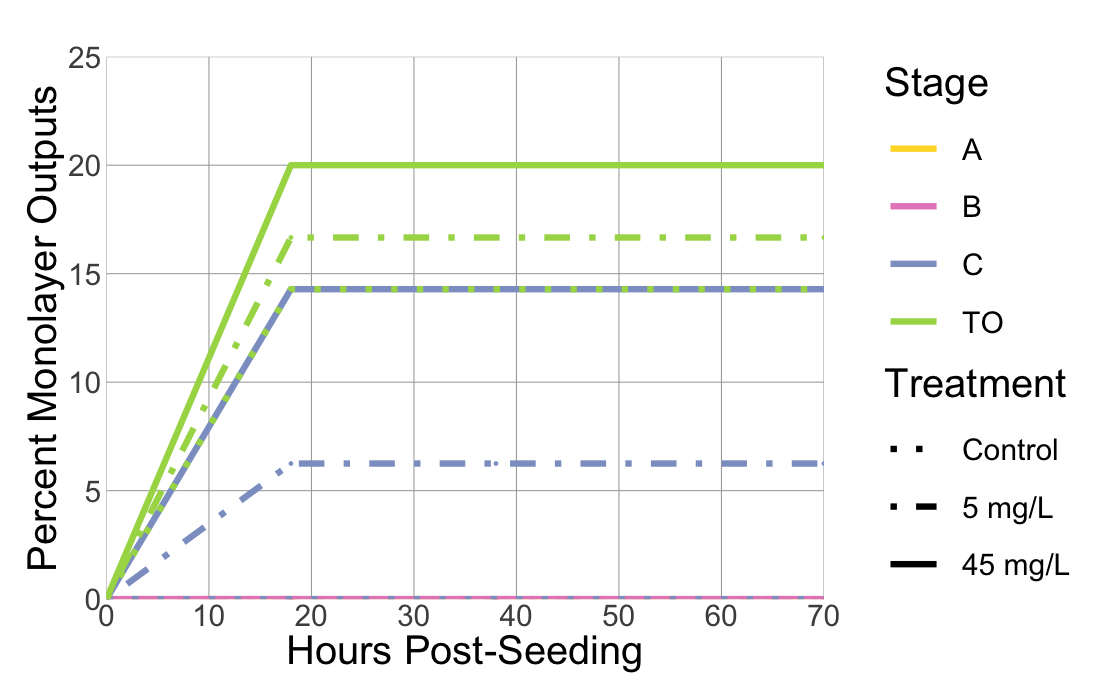
# png(filename = "line_percnet_output.png", width = 1100, height = 700) ##good for making a 7x5 in image for printing ~150 dpiggplot(data = mean_ratio_mono_out, aes(x = hps, y = percent_mean_mono, color = simple_stage, linetype = treatment_mg_per_L)) +theme_minimal() +labs(x ="Hours Post-Seeding", y ="Percent Monolayer Outputs") +geom_point() +geom_line(size =3) +# Adjusted line thickness to 2scale_color_manual(name ="Stage",values =c("#FFD92F", "#E78AC3", "#8DA0CB", "#A6D854"), # Specify the colorslabels =c("A", "B", "C", "TO")) +# Use fill instead of color in the legendscale_linetype_manual(name ="Treatment",values =c("dotted", "dotdash", "solid"), # Example linetypeslabels =c("Control", "5 mg/L", "45 mg/L")) +scale_x_continuous(breaks =seq(0, max(mean_ratio_mono_out$hps), by =10), expand =c(0, 0)) +# Adjust x-axis to start at 0scale_y_continuous(expand =c(0, 0), breaks =c(0, 5, 10, 15, 20, 25), limits =c(0, 25)) +# Adjust y-axis to start at 0theme(legend.position ="right",legend.margin =margin(1, 1, 1, 50), # Adjusting the margin around the legendaxis.text =element_text(size =30),axis.title =element_text(size =40),legend.text =element_text(size =30),legend.title =element_text(size =40),plot.margin =margin(t =2, r =1, b =1, l =1, unit ="cm"), # Adjust the top marginlegend.key.size =unit(4, "lines"),panel.grid.major.x =element_line(color ="darkgrey", size =0.5), # Remove major vertical gridlinespanel.grid.minor.x =element_blank(),panel.grid.major.y =element_line(color ="darkgrey", size =0.5),panel.grid.minor.y =element_blank()) # Adjust the size of legend keys for linetypes#dev.off()
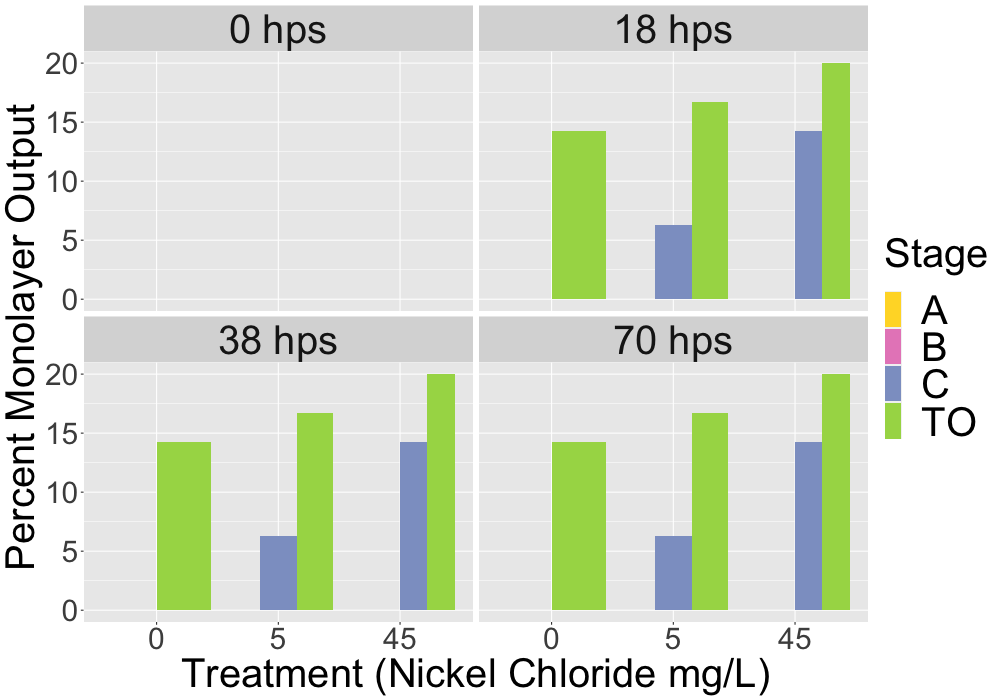
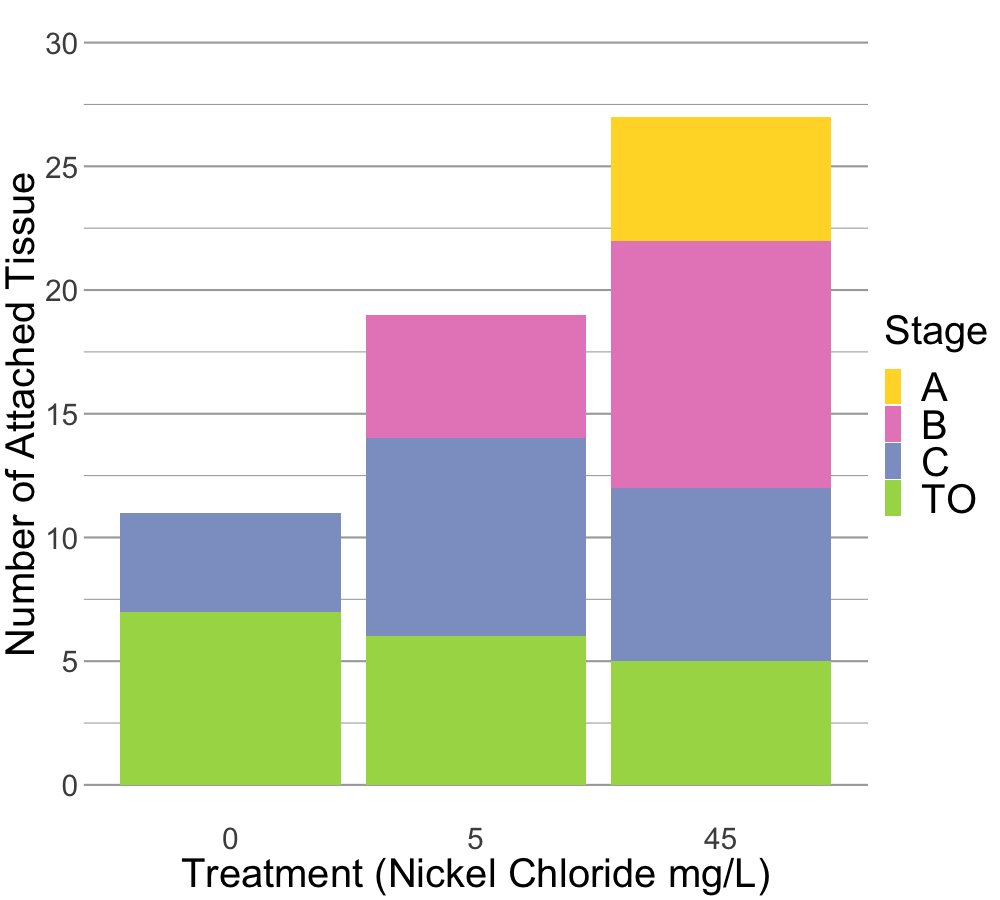
#png(filename = "mult_bar_plot_cell.png", width = 1000, height = 900) ## good for making a 3x3 in image for printing ~150 dpi# Create the ggplot with renamed hps values in facet_wrapggplot(mean_ratio_mono_out, aes(x = treatment_mg_per_L, y = percent_mean_mono, fill = stage_at_dissection)) +geom_bar(stat ="identity", position ="dodge", width =0.9) +scale_fill_brewer(palette ="Set2", name ="Stage") +labs(x ="Treatment (Nickel Chloride mg/L)",y ="Percent Monolayer Output") +facet_wrap(~ hps, labeller =as_labeller(c("18"="18 hps", "38"="38 hps", "70"="70 hps"))) +theme(axis.text=element_text(size=24),axis.title=element_text(size=30),legend.text =element_text(size =30),legend.title =element_text(size =30),strip.text =element_text(size =30)) # Adjusted facet label font size#dev.off()
#png(filename = "mult_bar_plot_cell_simple_stage.png", width = 1000, height = 700) # good for making a 3x3 in image for printing ~150 dpi# Create the ggplot with renamed hps values in facet_wrapggplot(mean_ratio_mono_out, aes(x = treatment_mg_per_L, y = percent_mean_mono, fill = simple_stage)) +geom_bar(stat ="identity", position ="dodge", width =0.9) +scale_fill_manual(name ="Stage",values =c("#FFD92F", "#E78AC3", "#8DA0CB", "#A6D854"), # Specify the colorslabels =c("A", "B", "C", "TO")) +labs(x ="Treatment (Nickel Chloride mg/L)",y ="Percent Monolayer Output") +facet_wrap(~ hps, labeller =as_labeller(c("0"="0 hps", "18"="18 hps", "38"="38 hps", "70"="70 hps"))) +theme(axis.text=element_text(size=30),axis.title=element_text(size=40),legend.text =element_text(size =40),legend.title =element_text(size =40),strip.text =element_text(size =40)) # Adjusted facet label font size#dev.off()
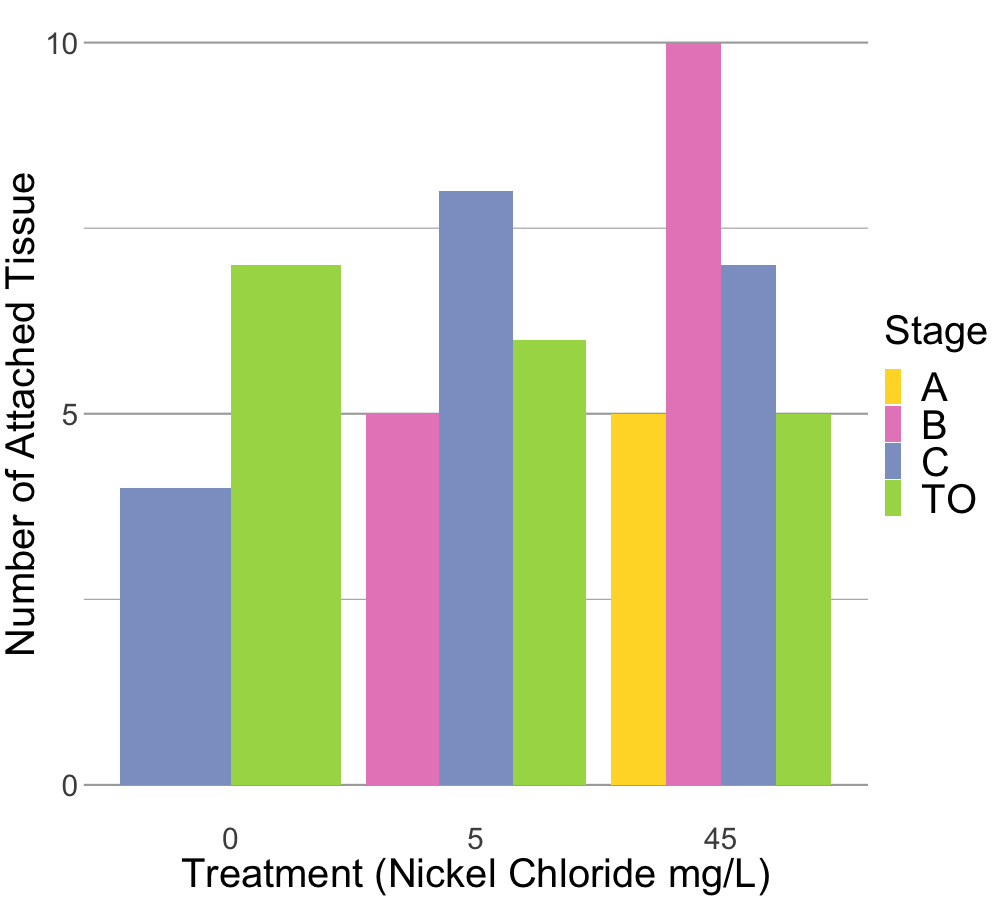
# would need to modify mean_ratio_tissue at top to include stage_at_dissection with the full range of stages if you need to modify this plot, currently color codes do not match with other plots available regarding stage.#png(filename = "cell_attached.png", width = 1000, height = 900)ggplot(mean_tissue_attached_18hps, aes(x = treatment_mg_per_L, y = mean_attached, fill = stage_at_dissection)) +geom_bar(stat ="identity", position ="dodge", width =0.9) +scale_fill_brewer(palette ="Set2", name ="Stage") +labs(x ="Treatment (Nickel Chloride mg/L)",y ="Number of Attached Tissue") +theme_minimal() +scale_y_continuous(breaks =c(0, 5, 10), limits =c(0, 10)) +theme(panel.grid.major.x =element_blank(), # Remove major vertical gridlinespanel.grid.minor.x =element_blank(),panel.grid.major.y =element_line(color ="darkgrey", size =1),panel.grid.minor.y =element_line(color ="darkgrey", size =0.5), axis.text =element_text(size =30),axis.title =element_text(size =40),legend.text =element_text(size =40),legend.title =element_text(size =40))#dev.off() # Close the PNG device
se <- (sd(cell_18hps$no_attached_tissue))/sqrt(length(cell_18hps$hps))sd(cell_18hps$no_attached_tissue)
[1] 1.972539
## Convert Treatment and HoursPostExposure to factors if they are not already factorscell_fact$treatment_mg_per_L <-as.factor(cell_fact$treatment_mg_per_L)cell_fact$hps <-as.factor(cell_fact$hps)## Perform two-way ANOVAanova_result <-aov(percent_output ~ treatment_mg_per_L + stage_at_dissection + treatment_mg_per_L:stage_at_dissection , data = cell_fact)# Summarize the ANOVA resultssummary(anova_result)
## Meeting the ANOVA assumptionsleveneTest(percent_output ~ stage_at_dissection*treatment_mg_per_L, data = cell_fact_filtered)
Levene's Test for Homogeneity of Variance (center = median)
Df F value Pr(>F)
group 10 0.6135 0.7912
33
#p-value > 0.05, therefore equal-variances are met# Check normality of residuals using Shapiro-Wilk testshapiro_test <-shapiro.test(residuals(anova_result))print(shapiro_test)
Shapiro-Wilk normality test
data: residuals(anova_result)
W = 0.79282, p-value = 8.977e-07
#DOES NOT meet assumption of normality
#================need to transform data, ===================## check for normality and homogenity again after this and rerun anovap_prime <-log(cell_fact_filtered$percent_output +1)cell_fact2 <-cbind(cell_fact_filtered, p_prime)var(cell_fact2$p_prime) # a ratio of less than 4 generally means you can assume equal variances as a rule of thumb
[1] 1.803502
leveneTest(p_prime ~ treatment_mg_per_L*stage_at_dissection, data = cell_fact2)
Levene's Test for Homogeneity of Variance (center = median)
Df F value Pr(>F)
group 10 0.6015 0.801
33
#p-value < 0.05, therefore equal-variances are not met again :( may be an issue with sample size## Perform two-way ANOVAanova_result2 <-aov(p_prime ~ treatment_mg_per_L + stage_at_dissection + treatment_mg_per_L:stage_at_dissection , data = cell_fact2)# Summarize the ANOVA resultssummary(anova_result2)
# Check normality of residuals using Shapiro-Wilk testshapiro_test <-shapiro.test(residuals(anova_result2))print(shapiro_test)
Shapiro-Wilk normality test
data: residuals(anova_result2)
W = 0.67709, p-value = 1.461e-08
# p less than 0.05, so my data is normally distributed now.## Perform Tukey's HSD test for significant interactiontukey_result <-TukeyHSD(x= anova_result2, ordered =TRUE, conf.level =0.95)print(tukey_result)
Tukey multiple comparisons of means
95% family-wise confidence level
factor levels have been ordered
Fit: aov(formula = p_prime ~ treatment_mg_per_L + stage_at_dissection + treatment_mg_per_L:stage_at_dissection, data = cell_fact2)
$treatment_mg_per_L
diff lwr upr p adj
5-0 0.34471457 -0.5393250 1.2287542 0.6088366
45-0 0.40041554 -0.4836241 1.2844551 0.5139450
45-5 0.05570096 -0.7627607 0.8741626 0.9847506
$stage_at_dissection
diff lwr upr p adj
B1-A2 9.436896e-16 -2.01699358 2.016994 1.0000000
B2-A2 5.570096e-02 -1.96129262 2.072695 0.9999994
C1-A2 2.280583e-01 -1.51870943 1.974826 0.9986436
C2-A2 1.484441e+00 -0.16242743 3.131309 0.0969222
TO-A2 2.312819e+00 0.66595066 3.959687 0.0021238
B2-B1 5.570096e-02 -1.96129262 2.072695 0.9999994
C1-B1 2.280583e-01 -1.51870943 1.974826 0.9986436
C2-B1 1.484441e+00 -0.16242743 3.131309 0.0969222
TO-B1 2.312819e+00 0.66595066 3.959687 0.0021238
C1-B2 1.723573e-01 -1.57441039 1.919125 0.9996512
C2-B2 1.428740e+00 -0.21812840 3.075608 0.1199378
TO-B2 2.257118e+00 0.61024969 3.903986 0.0028190
C2-C1 1.256383e+00 -0.04558108 2.558346 0.0636541
TO-C1 2.084761e+00 0.78279701 3.386725 0.0003932
TO-C2 8.283781e-01 -0.33613369 1.992890 0.2874634
$`treatment_mg_per_L:stage_at_dissection`
diff lwr upr p adj
5:C1-45:A2 5.956991e-16 -2.5180296 2.518030 1.0000000
45:B1-45:A2 1.023961e-15 -2.5180296 2.518030 1.0000000
5:B2-45:A2 1.268284e-15 -2.5180296 2.518030 1.0000000
0:C1-45:A2 1.497755e-15 -2.5180296 2.518030 1.0000000
0:C2-45:A2 1.719800e-15 -2.5180296 2.518030 1.0000000
5:C2-45:A2 1.952017e+00 -0.5660123 4.470047 0.2915768
0:TO-45:A2 2.045189e+00 -0.4728406 4.563219 0.2273221
45:C2-45:A2 2.045189e+00 -0.4728406 4.563219 0.2273221
5:TO-45:A2 2.153760e+00 -0.3642699 4.671789 0.1661741
45:TO-45:A2 2.283392e+00 -0.2346378 4.801421 0.1110225
0:A2-45:A2 NA NA NA NA
5:A2-45:A2 NA NA NA NA
0:B1-45:A2 NA NA NA NA
5:B1-45:A2 NA NA NA NA
0:B2-45:A2 NA NA NA NA
45:B2-45:A2 NA NA NA NA
45:C1-45:A2 NA NA NA NA
45:B1-5:C1 4.282622e-16 -2.5180296 2.518030 1.0000000
5:B2-5:C1 6.725851e-16 -2.5180296 2.518030 1.0000000
0:C1-5:C1 9.020562e-16 -2.5180296 2.518030 1.0000000
0:C2-5:C1 1.124101e-15 -2.5180296 2.518030 1.0000000
5:C2-5:C1 1.952017e+00 -0.5660123 4.470047 0.2915768
0:TO-5:C1 2.045189e+00 -0.4728406 4.563219 0.2273221
45:C2-5:C1 2.045189e+00 -0.4728406 4.563219 0.2273221
5:TO-5:C1 2.153760e+00 -0.3642699 4.671789 0.1661741
45:TO-5:C1 2.283392e+00 -0.2346378 4.801421 0.1110225
0:A2-5:C1 NA NA NA NA
5:A2-5:C1 NA NA NA NA
0:B1-5:C1 NA NA NA NA
5:B1-5:C1 NA NA NA NA
0:B2-5:C1 NA NA NA NA
45:B2-5:C1 NA NA NA NA
45:C1-5:C1 NA NA NA NA
5:B2-45:B1 2.443229e-16 -2.5180296 2.518030 1.0000000
0:C1-45:B1 4.737940e-16 -2.5180296 2.518030 1.0000000
0:C2-45:B1 6.958386e-16 -2.5180296 2.518030 1.0000000
5:C2-45:B1 1.952017e+00 -0.5660123 4.470047 0.2915768
0:TO-45:B1 2.045189e+00 -0.4728406 4.563219 0.2273221
45:C2-45:B1 2.045189e+00 -0.4728406 4.563219 0.2273221
5:TO-45:B1 2.153760e+00 -0.3642699 4.671789 0.1661741
45:TO-45:B1 2.283392e+00 -0.2346378 4.801421 0.1110225
0:A2-45:B1 NA NA NA NA
5:A2-45:B1 NA NA NA NA
0:B1-45:B1 NA NA NA NA
5:B1-45:B1 NA NA NA NA
0:B2-45:B1 NA NA NA NA
45:B2-45:B1 NA NA NA NA
45:C1-45:B1 NA NA NA NA
0:C1-5:B2 2.294711e-16 -2.5180296 2.518030 1.0000000
0:C2-5:B2 4.515157e-16 -2.5180296 2.518030 1.0000000
5:C2-5:B2 1.952017e+00 -0.5660123 4.470047 0.2915768
0:TO-5:B2 2.045189e+00 -0.4728406 4.563219 0.2273221
45:C2-5:B2 2.045189e+00 -0.4728406 4.563219 0.2273221
5:TO-5:B2 2.153760e+00 -0.3642699 4.671789 0.1661741
45:TO-5:B2 2.283392e+00 -0.2346378 4.801421 0.1110225
0:A2-5:B2 NA NA NA NA
5:A2-5:B2 NA NA NA NA
0:B1-5:B2 NA NA NA NA
5:B1-5:B2 NA NA NA NA
0:B2-5:B2 NA NA NA NA
45:B2-5:B2 NA NA NA NA
45:C1-5:B2 NA NA NA NA
0:C2-0:C1 2.220446e-16 -2.5180296 2.518030 1.0000000
5:C2-0:C1 1.952017e+00 -0.5660123 4.470047 0.2915768
0:TO-0:C1 2.045189e+00 -0.4728406 4.563219 0.2273221
45:C2-0:C1 2.045189e+00 -0.4728406 4.563219 0.2273221
5:TO-0:C1 2.153760e+00 -0.3642699 4.671789 0.1661741
45:TO-0:C1 2.283392e+00 -0.2346378 4.801421 0.1110225
0:A2-0:C1 NA NA NA NA
5:A2-0:C1 NA NA NA NA
0:B1-0:C1 NA NA NA NA
5:B1-0:C1 NA NA NA NA
0:B2-0:C1 NA NA NA NA
45:B2-0:C1 NA NA NA NA
45:C1-0:C1 NA NA NA NA
5:C2-0:C2 1.952017e+00 -0.5660123 4.470047 0.2915768
0:TO-0:C2 2.045189e+00 -0.4728406 4.563219 0.2273221
45:C2-0:C2 2.045189e+00 -0.4728406 4.563219 0.2273221
5:TO-0:C2 2.153760e+00 -0.3642699 4.671789 0.1661741
45:TO-0:C2 2.283392e+00 -0.2346378 4.801421 0.1110225
0:A2-0:C2 NA NA NA NA
5:A2-0:C2 NA NA NA NA
0:B1-0:C2 NA NA NA NA
5:B1-0:C2 NA NA NA NA
0:B2-0:C2 NA NA NA NA
45:B2-0:C2 NA NA NA NA
45:C1-0:C2 NA NA NA NA
0:TO-5:C2 9.317175e-02 -2.4248579 2.611201 1.0000000
45:C2-5:C2 9.317175e-02 -2.4248579 2.611201 1.0000000
5:TO-5:C2 2.017425e-01 -2.3162872 2.719772 1.0000000
45:TO-5:C2 3.313746e-01 -2.1866550 2.849404 1.0000000
0:A2-5:C2 NA NA NA NA
5:A2-5:C2 NA NA NA NA
0:B1-5:C2 NA NA NA NA
5:B1-5:C2 NA NA NA NA
0:B2-5:C2 NA NA NA NA
45:B2-5:C2 NA NA NA NA
45:C1-5:C2 NA NA NA NA
45:C2-0:TO 4.440892e-16 -2.5180296 2.518030 1.0000000
5:TO-0:TO 1.085707e-01 -2.4094589 2.626600 1.0000000
45:TO-0:TO 2.382028e-01 -2.2798268 2.756232 1.0000000
0:A2-0:TO NA NA NA NA
5:A2-0:TO NA NA NA NA
0:B1-0:TO NA NA NA NA
5:B1-0:TO NA NA NA NA
0:B2-0:TO NA NA NA NA
45:B2-0:TO NA NA NA NA
45:C1-0:TO NA NA NA NA
5:TO-45:C2 1.085707e-01 -2.4094589 2.626600 1.0000000
45:TO-45:C2 2.382028e-01 -2.2798268 2.756232 1.0000000
0:A2-45:C2 NA NA NA NA
5:A2-45:C2 NA NA NA NA
0:B1-45:C2 NA NA NA NA
5:B1-45:C2 NA NA NA NA
0:B2-45:C2 NA NA NA NA
45:B2-45:C2 NA NA NA NA
45:C1-45:C2 NA NA NA NA
45:TO-5:TO 1.296321e-01 -2.3883975 2.647662 1.0000000
0:A2-5:TO NA NA NA NA
5:A2-5:TO NA NA NA NA
0:B1-5:TO NA NA NA NA
5:B1-5:TO NA NA NA NA
0:B2-5:TO NA NA NA NA
45:B2-5:TO NA NA NA NA
45:C1-5:TO NA NA NA NA
0:A2-45:TO NA NA NA NA
5:A2-45:TO NA NA NA NA
0:B1-45:TO NA NA NA NA
5:B1-45:TO NA NA NA NA
0:B2-45:TO NA NA NA NA
45:B2-45:TO NA NA NA NA
45:C1-45:TO NA NA NA NA
5:A2-0:A2 NA NA NA NA
0:B1-0:A2 NA NA NA NA
5:B1-0:A2 NA NA NA NA
0:B2-0:A2 NA NA NA NA
45:B2-0:A2 NA NA NA NA
45:C1-0:A2 NA NA NA NA
0:B1-5:A2 NA NA NA NA
5:B1-5:A2 NA NA NA NA
0:B2-5:A2 NA NA NA NA
45:B2-5:A2 NA NA NA NA
45:C1-5:A2 NA NA NA NA
5:B1-0:B1 NA NA NA NA
0:B2-0:B1 NA NA NA NA
45:B2-0:B1 NA NA NA NA
45:C1-0:B1 NA NA NA NA
0:B2-5:B1 NA NA NA NA
45:B2-5:B1 NA NA NA NA
45:C1-5:B1 NA NA NA NA
45:B2-0:B2 NA NA NA NA
45:C1-0:B2 NA NA NA NA
45:C1-45:B2 NA NA NA NA
### still fails assumptions of homogenity for an anova so we need to try a non-parametric statistical test
Note that the sample size is so small that it fails to meet the assumptions of homogenity and a full statistical analysis of the data cannot be complete.